Qualification and Validation in Pharmaceutical Industry
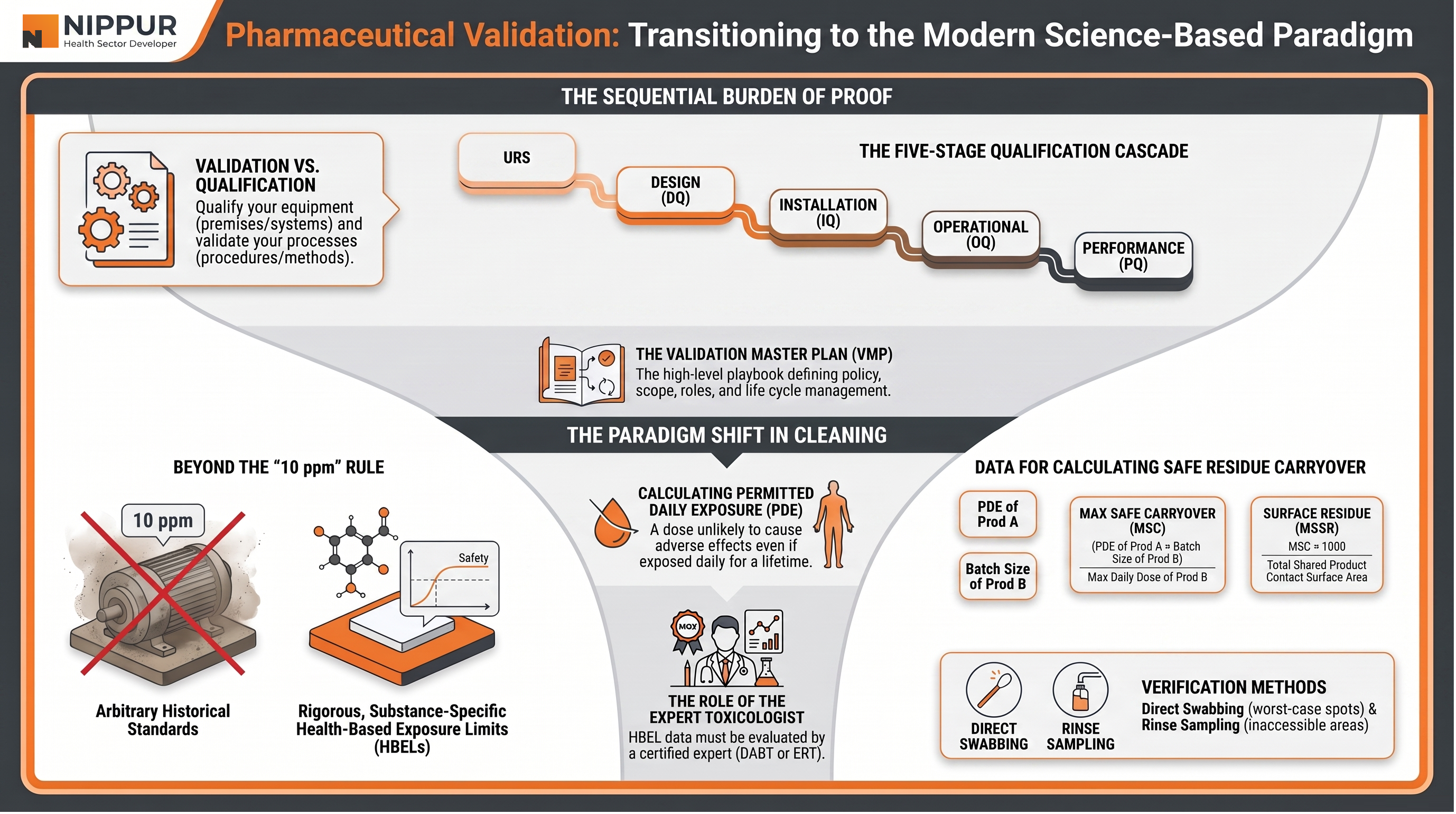
1. Defining the Foundation: Validation vs. Qualification
A common misconception in the industry is that validation and qualification are interchangeable; however, the WHO makes a distinct clarification:
- Validation: The documented evidence proving that a process, procedure, or method consistently leads to expected results.
- Qualification: The documented evidence that premises, systems, or equipment work correctly and are fit for their intended use.
All these activities are managed under the Validation Master Plan (VMP), the facility's "high-level playbook". The VMP establishes the overarching policy, defines the scope of what requires qualifying, assigns roles, and dictates lifecycle management for all systems.
2. The Equipment Qualification Lifecycle: The "Burden of Proof"
Qualification in the pharmaceutical industry follows a mandatory, sequential "burden of proof" that walks equipment from its conceptual stage to live production. Each stage must be successfully completed and documented before the next one can begin:
- User Requirement Specifications (URS): The process begins by defining exactly what the equipment is required to do.
- Design Qualification (DQ): This stage proves that the proposed design of the equipment actually meets the requirements laid out in the URS.
- Installation Qualification (IQ): This verifies that the equipment is bolted down, placed, and wired correctly according to specifications.
- Operational Qualification (OQ): Once installed, the equipment is tested to ensure it works correctly across all its intended operating ranges.
- Performance Qualification (PQ): The final step demonstrates that the equipment performs consistently and reliably under routine, real-world production conditions.
While the WHO allows for the combination of certain stages — such as a combined Installation and Operational Qualification (IOQ) for simpler machinery — the logical, sequential flow remains strictly mandatory.
3. Process Validation and the Lifecycle Approach
While qualification focuses on the hardware, process validation focuses on the procedure. Modern standards have shifted toward a continuous lifecycle approach consisting of three main stages:
- Stage 1: Process Design: Quality is designed and built into the process from day one.
- Stage 2: Process Qualification: Demonstrating that the process is capable of reproducible commercial manufacturing.
- Stage 3: Continued Process Verification: Using Statistical Process Control (SPC) to trend results and ensure the process remains in a validated state over its entire life cycle.
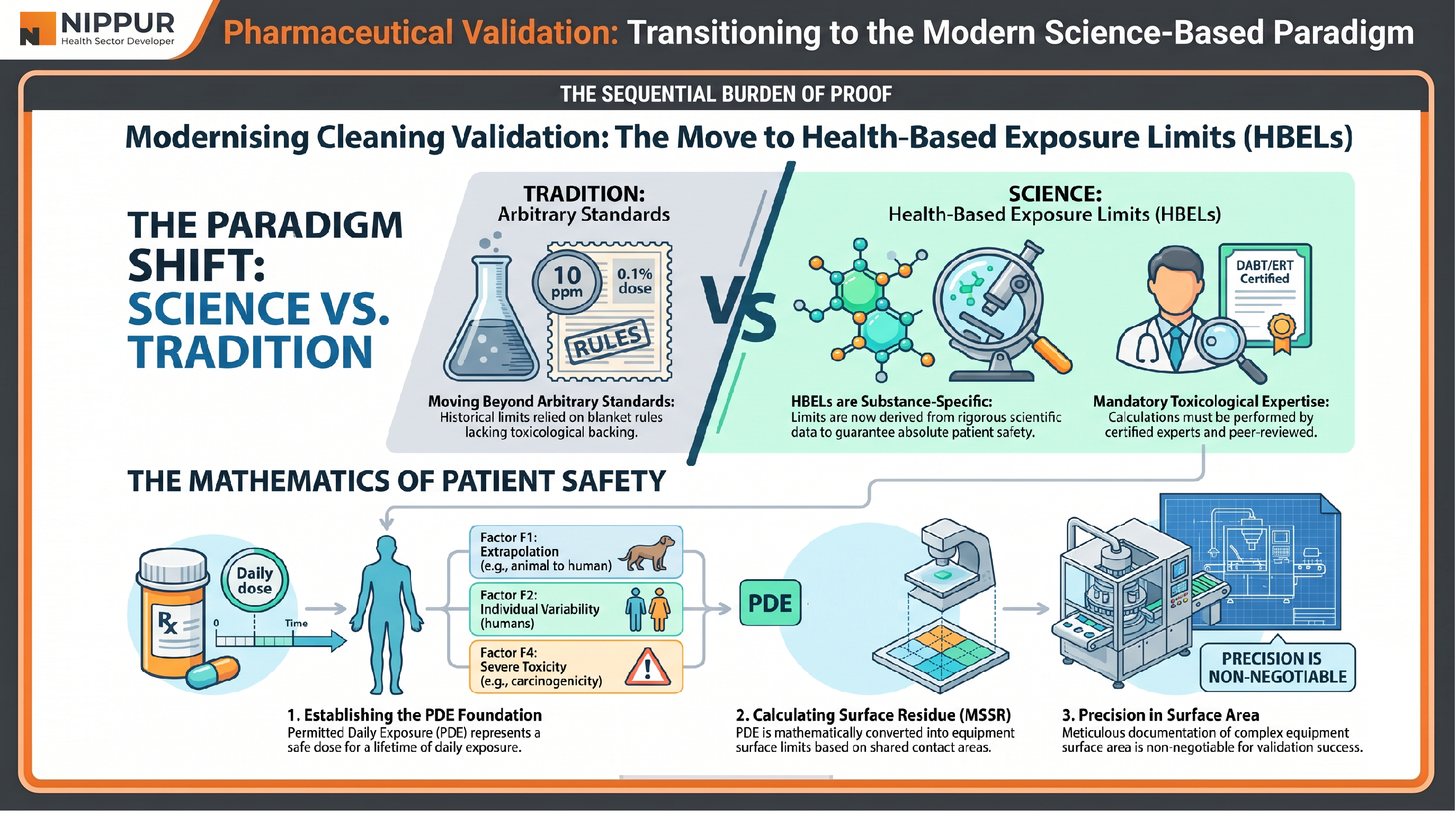
4. The Paradigm Shift in Cleaning Validation
One of the most critical aspects of validation is preventing cross-contamination in multi-product facilities. The industry is currently undergoing a massive shift from arbitrary historical limits to Health-Based Exposure Limits (HBELs).
For decades, industry relied on three basic criteria for cleaning validation:
- Visual cleanliness.
- The 10 ppm rule: no more than 10 parts per million of one product in another.
- The 0.1% dose rule: no more than 0.1% of a therapeutic dose in the next product's daily dose.
The World Health Organization (WHO) now mandates a shift to Health-Based Exposure Limits (HBELs). Unlike historical fractions, HBELs use rigorous, substance-specific toxicological data to ensure patient safety.
Calculating Safety: PDE, MSC, and MSSR
The foundational metric for this new safety standard is the Permitted Daily Exposure (PDE) — a dose unlikely to cause adverse effects even if exposed daily for a lifetime.
- PDE Calculation: Derived from the No Observed Adverse Effect Level (NOAEL) and adjusted by five factors (F1-F5) to account for species extrapolation, human variability, and toxicity severity.
- Carryover Limits: Once the PDE is established, facilities must calculate the Maximum Safe Carryover (MSC) and the Maximum Safe Surface Residue (MSSR).
- The Mathematical Link: There is a critical relationship between these limits and the physical surface area of the equipment. Accurate calculations for complex components like pumps and screens are non-negotiable for successful validation.
5. Data Integrity and Computerised Systems
In the digital age, qualification extends to software and automated systems. Following ALCOA+ principles, electronic records must be attributable, legible, contemporaneous, original, and accurate, as well as complete, consistent, enduring, and available.
Systems must feature independent computer-generated audit trails, secure time/date stamps, and unique user IDs to ensure that data cannot be altered or deleted.
By integrating these rigorous qualification stages and validation types into a unified quality system, the pharmaceutical industry ensures that every manufactured product meets the highest global safety standards.